+ Computational Protein, Enzyme, and DNA Design
+ Combinatorial Optimization & Protein Structure
+ Catalysis inside protein crystals
< Molecular Recognition for Synthetic Biology
Computational Modeling of Cytochrome P450 Specificity
+ Combinatorial Optimization & Protein Structure
+ Catalysis inside protein crystals
< Molecular Recognition for Synthetic Biology
Computational Modeling of Cytochrome P450 Specificity
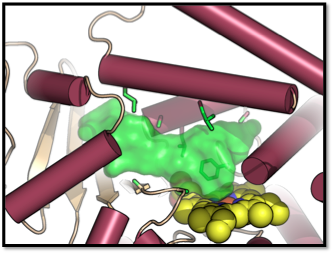
Cytochrome P450 enzymes (P450s) accept successive electrons at a heme bound iron atom which splits dioxygen and performs a variety of challenging chemical reactions. The Arnold group has used site-directed recombination to produce several thousand P450 enzymes from three parent sequences. About half (~3,000) of the designed P450s fold. We aim to develop new bioinformatics algorithms that mine this dataset to better understand P450 structure and stability. With detailed atomistic modeling and extensive conformational sampling, we will predict high resolution structures for libraries of P450s.
Every individual is chemically distinct, with various cytochrome P450 enzymes largely responsible for metabolism and clearance of therapeutic drugs. Few mammalian P450 crystal structures have been reported. However, recent evidence suggests that structure refinement of P450 homology models may now be feasible. We aim to pioneer computational methods to more accurately predict high resolution structures for libraries of cytochrome P450 enzymes. Structural models will be validated with experimental crystallography and spectroscopy. Modeling natural P450 sequences, allele variants, and designed P450s will yield a database of structures useful for understanding pharmacogenetic data and designing new therapeutic ligands.
One of the most visible early success stories for synthetic biology was the synthesis of an artemisinin precursor by Keasling and coworkers. This group was remarkable fortunate to extract the correct cytochrome P450 gene (CYP71AV1) from Artemisia annua. To reduce the need for luck in future synthetic biology endeavors, predictive models are needed for the substrate specificity of new cytochrome P450 genes. We aim to develop such predictive models, thereby organizing the natural P450 toolbox.