
Using Folding@Home volunteer computing power to identify better algorithms

In the near future we will release the first #N work units for Folding@Home beta testing. Most of the day, even when you are actively using your computer, only a fraction of the computing power is being used. By downloading the Folding@Home program, either the screensaver or the background-only version, you can donate the otherwise unused processing time to fundamental scientific research. Improved algorithms for high resolution protein structure prediction will be useful across the board, from engineering improved enzymes for biofuels to studying the molecular biophysics of biomedically relevant systems.
High resolution structure prediction
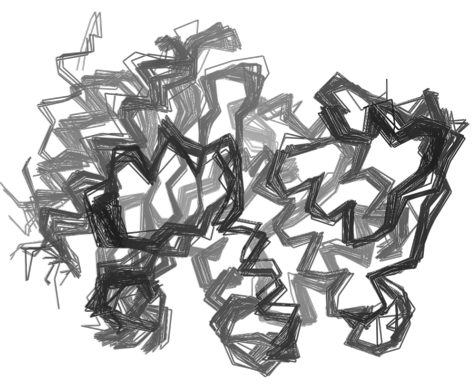
To better understand the chemistry that drives the fundamental processes of life, it is critical that we know the molecular structures of the many proteins involved in these processes. Unfortunately, our current laboratory techniques for determining protein structure, X-ray crystallography and NMR spectroscopy, are relatively slow, expensive and often difficult to perform.
Therefore, the creation of an automated tool for rapid protein structure determination would be nothing short of revolutionary. To this end, researchers have been working for several decades to design efficient algorithms to predict protein tertiary structure from the corresponding primary amino acid sequence. The obvious approach is to run a molecular dynamics simulation on an unfolded amino acid chain, but the amount of computational resources required makes this method intractable for all but the smallest proteins. Instead, the modern approach is to sample the protein's vast conformational space, looking for likely structures. An approximate Hamiltonian is computed for each, and the structure with the lowest energy is the result.
As an alternative to ab initio protein structure prediction (straight from an unfolded amino acid chain), it is often possible to start from an approximate homology model. In principle, one can then refine the structure until it is close to that of the real protein. Given the current state of available computing power, the limiting factor in this approach is conformational sampling; starting from a model that deviates roughly ~3 A from the true structure, the sheer number of similar structures that must be tested still makes most search strategies untenable.
Loop rebuilding
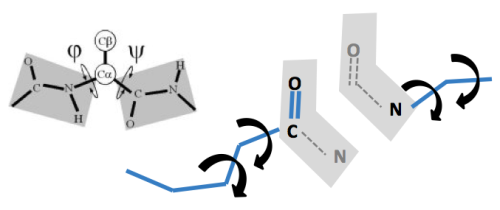
Loop rebuilding is an important problem in its own right. Leaders in this field include Jacobson, Friesner, Honig, Dunbrack and others. Simply put, the idea is this. Discard information for a protein segment of 3-12 residues. Now try to rebuild this segment. Traditionally this is called loop rebuilding because it is the loops (non alpha helical and non beta sheet residues) that are the target.
There are specialized sampling methods for building prospective loop coordinates. SHARPEN implements a variant of CCD (cyclic coordinate descent).
Hierarchical structure refinement
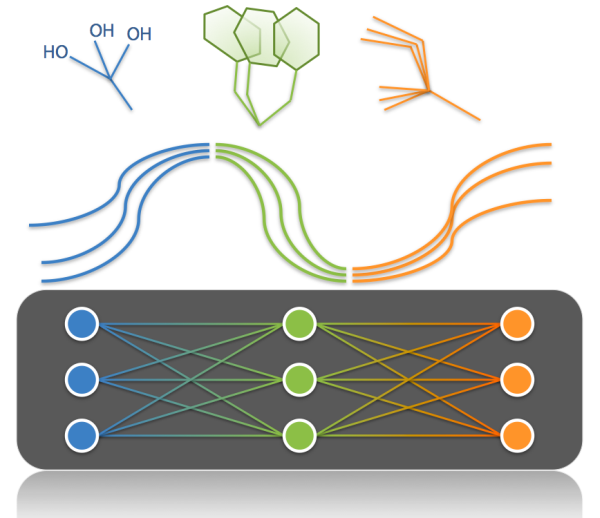
SHARPEN is an extensible platform for testing combinatorial optimization algorithms at the sidechain and backbone level on Folding@Home. There are special challenges associated with formulating protein structure refinement in terms of a discrete backbone search space, but benefits as well.